Current Research and Treatment
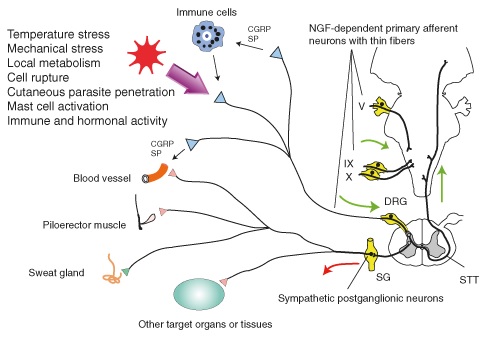
Figure 1: Many pathways stem from primary afferent neurons and
sympathetic post-ganglionic neurons.
Recent genetic studies of pain and analgesia have been helpful in uncovering the genetic basis of hereditary sensory and autonomic neuropathy type IV (HSAN IV), an autosomal recessive genetic disorder; in order for offspring to inherit the disease, both parents must possess and pass on the copy of the gene to the child. These studies have focused primarily on the nerve growth factor (NGF), a critical component for preserving primary afferent neurons and sympathetic post ganglionic neurons during embryonic development. NGF-dependent sensory neurons are also a basis of interoceptive sense, a homeostatic afferent pathway that transmits mechanical, thermal, chemical, metabolic, and hormonal information about the skin, joints, teeth, muscles, and vicera [1]. The absence of this pathway results in the inability to regulate favorable conditions for the smooth function of organs. For example, patients with HSAN IV are at the risk of organ failure from over-heating due to anhidrosis (stemming from the absence of sympathetic postganglionic neurons), the inability to sweat, which results from the inability of the body to detect temperature.
The Nerve Growth Factor
NGF, or, rather the lack thereof, was hypothesized to be the culprit behind HSAN IV because NGF-dependent neurons innervate all tissues of the body and, further, these neurons consist of myelinated Aδ-fibers and unmyelinated C-fibers, primary components of epicritic and protopathic pain pathways, respectively (Figure 1) [2, 3]. Since patients with HSAN IV have decreased intestinal motility and lack visceral pain perception, scientists have concluded that a subset of neurons comprising the glossopharyngeal nerve and the vagus nerve are also NGF-dependent [2]. During development, NGF is important for the suppression of apoptosis, cellular suicide; without NGF, primary afferent neuronal cells die [2].
Mutation in NGF-encoding Gene
The production of NGF begins with the synthesis of pro-form NGF (proNGF) and is followed by proteolytic processing before secretion as mature NGF. Therefore, mutations in the gene that encodes NGF, NGF beta (NGFB), are responsible for defective NGF and the resulting congential insensitivity to pain with anhidrosis and without anhidrosis [2]. One type of mutation, a missense mutation, discovered through the analysis of DNA from a family in northern Sweden revealed a substitution of the thymine nucleotide for cytosine nucleotide at position 661 and replacement of basic amino acid, arginine, by the non-polar amino acid tryptophan at position 221; this mutation results in only congenital insensitivity to pain (no anhidrosis) and the encoded proteins is mostly in the form of proNGF [4, 5]. Another type of mutation, loss-of-function mutation, observed in an Arab family in which five out of six children presented with no perception of pain, is characterized by a cytosine-to-adenine transversion at nucleotide 680 and a two nucleotide deletion at 681-681; this results in a frame-shift at amino acid valine 232 and the incorporation of 43 new amino acids in the place of 15; this mutation results in congenital insensitivity to pain, anhidrosis, and mental retardation [6]. Comparison of the two mutations suggests congenital insensitivity to pain and anhidrosis (as related to HSAN IV) to be associated with loss-of-function mutation. In summary, the complete defect of NGF (resulting from loss-of-function mutation), based on the comparison between the two cases, is linked to no perception of pain along with anhidrosis and mental retardation, while the missense mutation is linked to only congential analgesia.
Defective NGF Receptor: TrkA
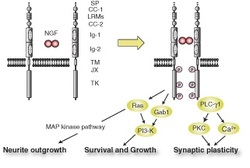
Figure 2: Pathways of TrkA
The action of NGF consists of activation of the tyrosine kinase A (TrkA) receptors at nerve terminals [2]. An activated TrkA is required for the potentiation of pain signals via various mechanisms. Three major intracellular signaling pathways are dependent on a phosphorylated TrkA: apoptosis, phosphatidyl inositol-3-kinase for suppressing apoptotic proteins, and mitogen-activated protein kinase for the axonal and dendrite outgrowth (Figure 2) [2, 4]. Mutations in the NTRK1 gene are the basis of defective TrkA receptors [4].
Treatment
Most treatment options available for HSAN IV are targeted at treating associated conditions. However, a drug by the name Naloxone, has been shown to reverse analgesia in some cases [7]. Originally used as an opioid receptor antagonist, Naloxone exerts the opposite effect of opioids on nociception. Low doses of Naloxone have been shown to lower the thermal and mechanical nociceptive threshold [8].
Treatment
Most treatment options available for HSAN IV are targeted at treating associated conditions. However, a drug by the name Naloxone, has been shown to reverse analgesia in some cases [7]. Originally used as an opioid receptor antagonist, Naloxone exerts the opposite effect of opioids on nociception. Low doses of Naloxone have been shown to lower the thermal and mechanical nociceptive threshold [8].
References
1. Lacroix-Fralish ML, Mogil JS. Progress in genetic studies of pain and analgesia. Annu Rev Pharmacol Toxicol. 2009;49:97–121.
2. Indo Y. Nerve growth factor and the physiology of pain: lessons from congenital insensitivity to pain with anhidrosis. Clinical Genetics. 2012; 82(4):341-350.
3. Basbaum AI, Bautista DM, Scherrer G, Julius D. Cellular and molecular mechanisms of pain. Cell. 2009; 139(2): 267–284.
4. Larsson E, Kuma R, Norberg A, Minde J, Holmberg M. Nerve growth factor R221W responsible for insensitivity to pain is defectively
processed and accumulates as proNGF. Neurobiology of Disease. 2009; 33(2): 221-228.
5. Einarsdottir E, Carlsson A, Minde J et al. A mutation in the nerve growth factor beta gene (NGFB) causes loss of pain perception. Hum Mol Genet. 2004;
13: 799–805.
6. Carvalho OP, Thornton GK, Hertecant J et al. A novel NGF mutation clarifies the molecular mechanism and extends the phenotypic spectrum of the HSAN5
neuropathy. J Med Genet. 2011; 48: 131–135.
7. Praveen KB, Sudhakar S, Prabhat MPV. Congenital insensitivity to pain. Online J Health Allied Scs. 2010; 9(4): 29.
8. Hafeshiani ZK, Karami M, Biglarnia M. Nitric oxide in hippocampal cortical area interacts with naloxone in inducing pain. Indian J Pharmacol. 2012; 44(4):
443–447.
2. Indo Y. Nerve growth factor and the physiology of pain: lessons from congenital insensitivity to pain with anhidrosis. Clinical Genetics. 2012; 82(4):341-350.
3. Basbaum AI, Bautista DM, Scherrer G, Julius D. Cellular and molecular mechanisms of pain. Cell. 2009; 139(2): 267–284.
4. Larsson E, Kuma R, Norberg A, Minde J, Holmberg M. Nerve growth factor R221W responsible for insensitivity to pain is defectively
processed and accumulates as proNGF. Neurobiology of Disease. 2009; 33(2): 221-228.
5. Einarsdottir E, Carlsson A, Minde J et al. A mutation in the nerve growth factor beta gene (NGFB) causes loss of pain perception. Hum Mol Genet. 2004;
13: 799–805.
6. Carvalho OP, Thornton GK, Hertecant J et al. A novel NGF mutation clarifies the molecular mechanism and extends the phenotypic spectrum of the HSAN5
neuropathy. J Med Genet. 2011; 48: 131–135.
7. Praveen KB, Sudhakar S, Prabhat MPV. Congenital insensitivity to pain. Online J Health Allied Scs. 2010; 9(4): 29.
8. Hafeshiani ZK, Karami M, Biglarnia M. Nitric oxide in hippocampal cortical area interacts with naloxone in inducing pain. Indian J Pharmacol. 2012; 44(4):
443–447.
