Pathophysiology
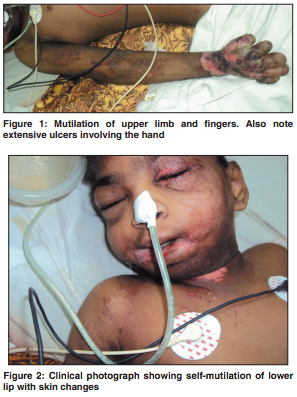
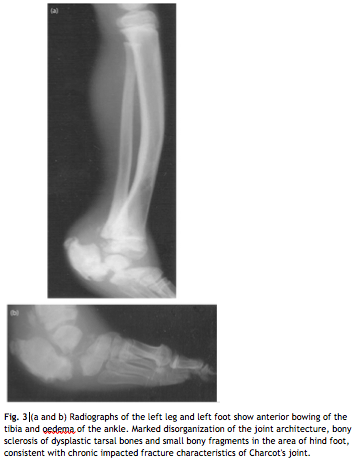
Hereditary sensory and autonomic neuropathy type IV is an autosomal disease seen in children with several clinicial indications to look out for. It is a very rare condition with less than 50 cases reported up to 2006. (1) First, a history of self mutilation in infancy after dentition is a symptom of HSAN IV as a result of poor pain perception, and the levels of this vary by patient. Anhidrosis can be observed during hot weather with high fevers and in noticeably dry skin, due to poor temperature perception and the loss of the ability to sweat. (2) Mental retardation and sever learning disorders are often present. Upon further examination of the patient’s neurology, HSAN IV patients have a highly reduced ability to respond to temperature and pain stimuli. For example, patients often experience bone breaks or changes and experience no pain as a result. (1) These conditions point out the possible presence of HSAN IV. The factor that differentiates HSAN IV from HSAN I, HSAN II, HSAN III, and HSAN V is the negative sweat test. All other aspects of the child’s development are normal, with typical tendon and plantar reflexes.
|
|
Genetic Mutations
Genetic mutations in the SCN9A and NTRK1 gene have been linked to the diagnosis of HSAN-IV/CIPA.
The SCN9A gene codes for one of the 9-united family of voltage-gated sodium ion channels, Na1.7. [3] Previously discussed, voltage-gated sodium and potassium channels are responsible for the firing of action potentials in neurons because they facilitate the flow of sodium and potassium ions down their electrochemical gradient. While other sodium channels in the Na1.1-Na1.9 family are located on specific neurons and neuron fibers, the Na1.7 ion channels are found in all neurons, and especially in abundance on nociceptors, the sensory neurons responsible for detecting pain.[4] Proper gene expression of SCN9A gene is essential to action potential behavior, as the Na1.7 ion is first to fire during an action potential, eventually signaling the opening of the Na1.8 voltage gated channel, located on C-fibers, the nerve bundle responsible for carrying pain signals to the central nervous system. [5]. Mutations in the SCN9A gene are divided into loss-of-function mutations or gain-of-function mutations. In gain of function mutations, the resulting Na1.7 ion channel encoded by the gene causes the hyperactivity of Na+ ion transport. As a result, gain-of-function mutations, usually nonsense mutations[6], are the causes of extreme pain or burning sensations, as well as inability to stop feeling pain. Loss-of-function mutations in Na1.7 voltage gated ion channels result in no activity in the encoded ion channel and as a result, no Na+ ion flow. Because of the role of Na1.7 in signal transduction to C-fibers from nociceptors (and in neurons in general) a pain signal will not be detected from the C-fibers or CNS due to the lack of action potentials firing.
Genetic mutations involving the NTRK1 gene have been directly connected to the diagnosis of HSAN-IV. [7] The NTRK1 gene codes for growth factors that help the development of neurons, especially nocicpetors and temperature sensory neurons. Typically located on the surface of the cells, neurotrophic tyrosine kinase, receptor, type 1, phosphorylates itself when bound by the protein nerve growth factor beta. The phosphorylation acts as an on switch which induces signals for neuron growth. Mutations in the NTRK1 gene in turn may code for a receptor that is unable to phosphorylate itself, thus unable to phosphorylate other proteins needed for neuron growth.[8] As a result, neurons, especially nociceptors, die by cell apoptosis. Insensitivity to pain, then, would steam from the fact that there would be no living nociceptors in the body. In addition, the NTRK1 gene also codes for proteins that aid in neural differentiation, in other words, NTRK1 helps in differentiating the different type of neural sensory receptors. [9] Genetic mutations in the the gene could result in error in the proteins used to differentiate different sensory neurons. As a result, a wrong protein may change the intended function of the sensory neuron.
The SCN9A gene codes for one of the 9-united family of voltage-gated sodium ion channels, Na1.7. [3] Previously discussed, voltage-gated sodium and potassium channels are responsible for the firing of action potentials in neurons because they facilitate the flow of sodium and potassium ions down their electrochemical gradient. While other sodium channels in the Na1.1-Na1.9 family are located on specific neurons and neuron fibers, the Na1.7 ion channels are found in all neurons, and especially in abundance on nociceptors, the sensory neurons responsible for detecting pain.[4] Proper gene expression of SCN9A gene is essential to action potential behavior, as the Na1.7 ion is first to fire during an action potential, eventually signaling the opening of the Na1.8 voltage gated channel, located on C-fibers, the nerve bundle responsible for carrying pain signals to the central nervous system. [5]. Mutations in the SCN9A gene are divided into loss-of-function mutations or gain-of-function mutations. In gain of function mutations, the resulting Na1.7 ion channel encoded by the gene causes the hyperactivity of Na+ ion transport. As a result, gain-of-function mutations, usually nonsense mutations[6], are the causes of extreme pain or burning sensations, as well as inability to stop feeling pain. Loss-of-function mutations in Na1.7 voltage gated ion channels result in no activity in the encoded ion channel and as a result, no Na+ ion flow. Because of the role of Na1.7 in signal transduction to C-fibers from nociceptors (and in neurons in general) a pain signal will not be detected from the C-fibers or CNS due to the lack of action potentials firing.
Genetic mutations involving the NTRK1 gene have been directly connected to the diagnosis of HSAN-IV. [7] The NTRK1 gene codes for growth factors that help the development of neurons, especially nocicpetors and temperature sensory neurons. Typically located on the surface of the cells, neurotrophic tyrosine kinase, receptor, type 1, phosphorylates itself when bound by the protein nerve growth factor beta. The phosphorylation acts as an on switch which induces signals for neuron growth. Mutations in the NTRK1 gene in turn may code for a receptor that is unable to phosphorylate itself, thus unable to phosphorylate other proteins needed for neuron growth.[8] As a result, neurons, especially nociceptors, die by cell apoptosis. Insensitivity to pain, then, would steam from the fact that there would be no living nociceptors in the body. In addition, the NTRK1 gene also codes for proteins that aid in neural differentiation, in other words, NTRK1 helps in differentiating the different type of neural sensory receptors. [9] Genetic mutations in the the gene could result in error in the proteins used to differentiate different sensory neurons. As a result, a wrong protein may change the intended function of the sensory neuron.
References
- Marik, Ivo, Miroslav Kuklik, Dana Kuklikova, and Kazamierz Kozlowsk. "Hereditary Sensory and Autonomic Neuropathy Type IV Orthopaedic Complications."Journal of Pediatric Orthopaedics B 18 (2009): 138-40. Wolters Kluwer. Web. 29 Nov. 2012.
- Prashanth, G.P., and Mahesh Kamete. "A Case of Hereditary Sensory Autonomic Neuropathy Type IV." Ann Indian Acad Neurol 15 (2012): 134-36. EBSCOhost. Web. 29 Nov. 2012. <http://web.ebscohost.com/ehost/pdfviewer/pdfviewer?sid=14ead202-362e-4ca3-a233-599d55e106f7%40sessionmgr113&vid=2&hid=119>.
- Axelrod, Felicia B. "Hereditary Sensory And Autonomic Neuropathies: Types II, III, And IV." Orphanet Journal Of Rare Diseases 2.(2007): 39-50. Academic Search Complete. Web. 26 Nov. 2012.
- Klein, Christoper J, and Yanhong Wu. “Infrequent SCN9A Mutations in Congenital Insensitivity to Pain and Erythromelalgia.” Neurogenetics. 11(2012): 1-6. Academic Search Completel. Web. 23 Nov. 2012.
- Reimann, F. "Pain perception is altered by a nucleotide polymorphism in SCN9A." PNAS 2.(2010): 5148-5153. Academic Search Complete. Web. 26 Nov. 2012
- Liu, Min, and John N. Wood. "The Roles Of Sodium Channels In Nociception: Implications For Mechanisms Of Neuropathic Pain." Pain Medicine 12.(2011): S93-S99. Academic Search Complete. Web. 26 Nov. 2012.
- Axelrod FB, Gold-von Simson G, Oddoux C. Hereditary Sensory and Autonomic Neuropathy IV. 2008 Aug 5 [Updated 2009 Nov 24]. In: Pagon RA, Bird TD, Dolan CR, et al., editors. GeneReviews™ [Internet]. Seattle (WA): University of Washington, Seattle; 1993-.Available from: http://www.ncbi.nlm.nih.gov/books/NBK1769/
- "NTRK1 Neurotrophic Tyrosine Kinase, Receptor, Type 1 [ Homo Sapiens ]." Gene. N.p., 25 Nov. 2012. Web. 26 Nov. 2012. <http://www.ncbi.nlm.nih.gov/gene/4914#summary>.
- "NTRK1." Genetics Home Reference. US National Library of Medicine, 25 Nov. 2012. Web. 26 Nov. 2012. <http://ghr.nlm.nih.gov/gene/NTRK1>.
